
单克隆抗体的亲和力的筛选及优化对候选药物的开发至关重要,因为它可以影响药物的疗效,从而影响剂量和给药方案。本研究聚焦于优化一种创新的“二合一”抗体,该抗体能够同时靶向表皮生长因子受体(EGFR)和程序性死亡配体1(PD-L1),由于替换了轻链CDR3区域的单个氨基酸,分离的抗体变体靶向EGFR的亲和力提高了60倍。同时使用多种方法确认了二合一变体的结合特性,包括应用BLI、switchSENSE®及原生分子互作细胞分析系统(RT-ICRT-IC)等方法进行验证。结合实验研究团队通过使用位点定向突变和酵母表面展示技术,成功地对EGFR结合位点进行了亲和成熟。该研究不仅展示了抗体工程在提高药物疗效方面的潜力,还为未来的癌症治疗提供了新的思路。
在过去几十年中,单克隆抗体(mAbs)已成为治疗各种疾病的重要药物类别。为了提高疗效并减少副作用,抗体对其抗原具有高亲和力和特异性至关重要[1]。抗体的抗原结合位点由6个互补决定区(CDRs)组成,其中3个位于重链上,3个位于轻链上[2]。在抗体亲和力成熟过程中,CDRs经历了高度的体细胞突变。在哺乳动物中,通过免疫球蛋白(Ig)基因的突变多样性产生高亲和力抗体,这一过程称为体细胞超突变(SHM)[3,4]。
由于Ig基因的随机SHM和含有亲和力增强突变的B细胞的选择及克隆扩增之间的交替过程,在免疫反应中抗体的亲和力大幅提升[5]。治疗性抗体可以来源于多种物种,如小鼠、大鼠、家兔、鸡或非人灵长类动物[6,7]。动物免疫的优势在于抗体已经通过几轮SHM在体内经历了亲和力成熟[8]。然而,即使是体内衍生的抗体也可能没有对抗原所需的亲和力,这随后可以通过体外亲和力成熟进行优化[9]。为此,可以应用随机或有针对性的突变[8]。
靶向诱变方法通过定点诱变产生抗体变体库,突变可设计CDR区域[8,10]。可通过展示淘选技术(如噬菌体展示[11]、酵母表面展示[12]或核糖体展示[13-14])进行亲和筛选来分离最佳成熟的抗体变体。但对于包含所有可能单突变和多突变组合的抗体库,其规模可能超1030个变体,难以用常规展示技术筛选,而热点诱变、透视诱变和同步诱变可提高CDR多样化效率并减小库的规模[15-17]。
对于单特异性抗体,优化亲和力可影响药物疗效、剂量和给药方案,限制不良反应并降低治疗成本。而双特异性抗体(bsAbs)在癌症治疗中具有独特优势,能够交联受体或阻断两种疾病相关的信号通路,通过同时靶向同一恶性细胞上的两种癌症特异性抗原,可增强肿瘤特异性[18-20]。在许多实体瘤中,EGFR和PD-L1是两种重要的治疗靶点 [21,22]。该研究针对一种同时靶向EGFR和PD-L1的Two-in-One抗体(HCP-LCE)展开。这种抗体通过对两只鸡分别用EGFR和PD-L1胞外域免疫,然后将各自的重链和轻链模块组合成Fab格式以分离同时结合两个靶点的抗体变体[23]。HCP-LCE通过结合二聚化结构域II抑制EGFR信号传导,并阻断PD-1/PD-L1相互作用,对每个抗原具有中等个体亲和力。
该研究旨在探究能否在不丧失对PD-L1结合能力的前提下,优化该抗体对EGFR的亲和力,这是首次对动物免疫和组合筛选获得的Two-in-One抗体进行亲和力成熟研究,通过位点定向诱变和酵母表面展示(YSD)结合荧光激活细胞分选(FACS)优化 HCP-LCE对EGFR的亲和力。
为构建酵母文库,该研究设计针对轻链CDR1(LCDR1)和CDR3(LCDR3)特定氨基酸的引物,通过PCR反应构建文库。将VL基因通过同源重组转入酵母载体,与HCP-LCE的重链结合进行Fab展示。诱导基因表达后,用不同浓度的EGFR-Fc等进行孵育,再用相应抗体检测,最后通过FACS筛选。
对筛选出的酵母载体测序,将VL片段重新格式化到pTT5衍生载体,转染Expi293FTM细胞表达全长抗体,收集细胞培养上清,用MabSelectTM PrismA HP柱纯化,再进行缓冲液交换。
Expi293FTM细胞在Expi293TM表达培养基中培养,每3-4天传代一次,37℃、8% CO2孵育。A431和A549细胞在T75细胞培养瓶中,在37℃和5% CO2条件下,在Dulbecco 's Eagle培养基中添加10% FBS和1%青霉素-链霉素中培养,每3-4天传代一次。Jurkat细胞保存在添加10% FBS和1%青霉素-链霉素的RPMI-1640中,每3-4天传代一次,37℃和5% CO2孵育。
在CFX Connect Real-Time PCR Detection System中,设置温度梯度,测定抗体变体在不同温度下的稳定性,计算熔解温度(Tm)。
使用显示EGFR-ECD截断版本的酵母细胞在亚结构域水平上进行EGFR表位定位。与相应抗体孵育,用FITC共轭抗-c-myc抗体和抗人IgG Fc PE共轭抗体分别验证表面展示和抗体结合,通过流式细胞术(CytoFLEX S系统)分析细胞。
0
6用Octet RED96系统,将HCP-LCE变体固定在抗人IgG-Fc捕获生物传感器上,分别与不同浓度的EGFR-ECD和PD-L1-ECD孵育,测定亲和力和结合动力学参数。用ForteBio数据分析软件9.0.0.14分析数据,采用Savitzky-Golay过滤和1:1 Langmuir结合模型确定结合动力学参数。
在PD-1竞争实验中,将HCP-LCE和LCE-E固定在抗人Fab-CH1第二代生物传感器上。为了进行同时结合实验,AHC生物传感器上装载了单臂(oa)版本的HCP-LCE或LCE-E。作为对照,oaHCP-LCE和oaLCE-E仅与PD-L1孵育。
switchSENSE®测量在helix+仪器(Dynamic Biosensors,Munich, Germany)中进行,使用标准适配器芯片(ADP-48-2-0, Dynamic Biosensors)在静态荧光接近传感(FPS)模式下进行。在准备测量时,使用heliX®胺偶联试剂盒1 (HK-NHS-1, Dynamic Biosensors)将目标蛋白偶联到配体链上。蛋白-DNA偶联物在proFIRE®(Dynamic Biosensors)中纯化。两个蛋白-DNA偶联物被分离。实验采用较早的洗脱共轭物(共轭物1)。PD-L1配体链偶联物与适配器链1用红色染料Ra (AS1-Ra, Dynamic Biosensors)混合,EGFR配体链偶联物与适配器链1用绿色染料Ga (AS1-Ga, Dynamic Biosensors)混合20 min,温度25℃。
对于单蛋白表面实验,20% PD-L1-LS/AS1-Ra或20% EGFR-LS/AS1-Ga与80%互补锚链1 (c-Anchor 1, DC-0, Dynamic Biosensors)混合。对于双蛋白表面实验,电极1的功能化混合物含有10% PD-L1-LS/AS1-Ra, 10% EGFR-LS/AS1-Ga和80%互补锚链1 (c-Anchor 1, DC-0, Dynamic Biosensors)。电极2的功能化混合物包含无配体链(LFS-0,Dynamic Biosensors)与具有相应染料(AS-2-Ra和AS-2-Ga,Dynamic Biosensors)的适配器链2混合,并以相应比例与c-锚2 (DC-0,Dynamic Biosensors)混合。工作流在heliOS软件v2024.1.0中设置。表面被功能化了200秒。从10倍浓缩缓冲液(BU-PE-140-10, Dynamic Biosensors)中稀释1倍PE140,将分析物稀释至1E-8 M, 1E-9 M和1E-10 M,作为运行缓冲液。以200 µL/min的流速测定180 s的关联。以500 µL/min的流速测定1800s的解离。在仅PD-L1的实验中,红色LED功率设置为2%,绿色LED功率设置为0%。
在EGFR-only实验中,绿色LED功率设置为2%,红色LED功率设置为0%。在双蛋白/双色实验中,两个led都使用2%的功率。测量在25°C下进行。用heliOS软件v2024.1.0对测得的结合曲线进行评价。所得的双参考数据点采用连续振幅的单相结合-双相解离模型进行拟合。使用R package ggplot2绘制拟合曲线。
在将A549细胞与不同浓度的抗体孵育,再用抗人IgG Fc PE共轭抗体检测,通过流式细胞术测定细胞结合情况,所得曲线采用GraphPad Prism进行参数拟合。
在芯片型号为M5 (CY-M5-1, Dynamic Biosensors)的heliXcyto装置(Dynamic Biosensors)上测量细胞的实时动力学。抗体用红色染料标记,使用heliXcyto标记试剂盒red dye 1 (CY-LK-R1-1, Dynamic Biosensors),按照说明书进行标记。用光度法测定标记度(DOL)。HCP-LCE和LCE-E的测定DOL分别为2.5和4。将细胞重悬于PBS中,并通过30 µm细胞滤器(FIL-30-20, Dynamic Biosensors)进行筛选。除了A431细胞上的LCE-E的一次测量外,所有的相互作用都是在室温下用2% PFA处理15分钟的细胞上测量的。细胞以2×106个/mL的浓度捕获。抗体HCP-LCE和LCE-E分别稀释至60/20/6.67 nM和100/50/25 nM。使用从10倍原液(BU-RB-10-1, Dynamic Biosensors)稀释的1×PPBS进行抗体稀释,并作为运行缓冲液。工作流程在heliOS软件v2024.1.0中设置。用相应浓度的红色染料(NOR-0, Dynamic Biosensors)的归一化溶液对信号进行归一化处理。红色LED功率设置为0.11-0.13%。测量在25℃下进行,自动进样器冷却至4℃。测定每一种结合作用300 s,测定每一种解离作用1800 s。利用heliOS软件v2024.1.0对测得的结合曲线进行评价。根据归一化溶液得到的信号,对Spot 1和Spot 2的结合曲线进行归一化处理。使用R package ggplot2绘制并拟合曲线。
用ColabFold版本1.5.5进行建模[24-27],MMseq2生成多个序列比对(msa)[28,29],使用UniRef100数据库[30]并通过AMBER力场松弛模型[31],用PRODIGY计算结合能和解离常数[32]。EGFR使用AlphaFold2对接,随后利用HDOCK对接PD-L1[33]。
通过筛选鸡源酵母表面展示文库确定二合一抗体HCP- LCE的EGFR结合模块(图1A),该文库由抗PD-L1抗体重链与免疫抗EGFR轻链文库结合而成。因HCP-LCE同一Fv 区同时靶向EGFR和PD-L1且PD-L1结合主要靠重链CDR,所以认为轻链CDR主要参与EGFR结合[23]。
为使 HCP-LCE的EGFR结合亲和力成熟,随机化 LCDR1 和 LCDR3 的单氨基酸生成YSD文库(图1B),未对LCDR2修饰。YSD文库设计为LCDR1的9个氨基酸、LCDR3的12个氨基酸各含一个突变,每个轻链最大突变率为2个突变,采用位点饱和诱变[34],用退化的NNS密码子减少终止密码子(图1B),组合所有可能突变对,理论文库大小在蛋白和DNA水平分别为4.3×10⁴个轻链突变体和1.1×10⁵个突变体。NNS YSD文库克隆时,VL片段经同源重组插入载体,再通过酵母交配与编码野生型二合一 VH-CH1片段的酵母细胞结合(图1B)。
二倍体二合一 LC NNS文库经 FACS 筛选高亲和力EGFR结合物,共三轮(图1C),从500 nM EGFR-fc开始。第二轮筛选进行竞争筛选,用100 nM 标记的EGFR与500 nM野生型抗体预孵育对富集文库染色(图1C),双阳性酵母细胞表明其靶向EGFR的亲和力高于HCP-LCE。第三轮分选后,对12个克隆的VL片段测序检测EGFR和PD -L1结合。
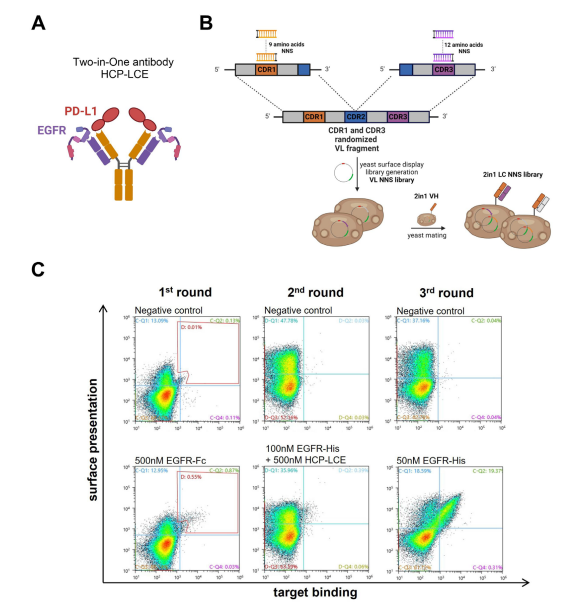
图1. HCP-LCE轻链NNS 酵母表面展示文库的生成和筛选。(A)EGFR和PD-L1结合的二合一抗体HCP-LCE的示意图。(B)YSD文库生成的克隆步骤示意图。LCDR1和LCDR3的单个氨基酸被编码20种天然氨基酸的密码子所取代。通过酵母交配将轻链多样性与野生型HCP-LCE重链结合。使用BioRender.com创建。(C)通过流式细胞术对二倍体 HCP-LCE 轻链突变 YSD文库进行筛选。y轴表示利用抗人λ链抗体 AF647或PE标记的表面展示,而X轴分别利用抗人Fc PE抗体或抗6×His AF647抗体表示EGFR-Fc或EGFR-His结合。
分析的12个二合一的LC突变体中,多数有野生型 LCDR1 区域,这表明轻链CDR1 并非主要参与EGFR结合;所有变异体在 LCDR3 同一位置突变,显示该位置对EGFR 结合有主要影响,12突变体中,有11个在LDCR3的第3位酪氨酸向谷氨酸突变,也有1有向天冬氨酸突变的。还产生一个携带酪氨酸到赖氨酸突变的变体。
Expi293FTM 细胞被共转染产生全长二合一抗体突变体,突变体经蛋白A亲和层析纯化。SDS-PAGE分析显示有预期的重链及轻链且无降解产物,热稳定性研究表明HCP-LCE变体熔化温度在60.5℃至66.5℃之间,野生型抗体的熔化温度为58.9℃,表明热稳定性没有降低(表1)。
表1. HCP-LCE、LCE-E、LCE-D和LCE-K的生物物理性质,包括BLI测量的亲和力和动力学结合率、流式细胞术测量的EC50值、PRODIGY web服务器预测的亲和力值和熔化温度。
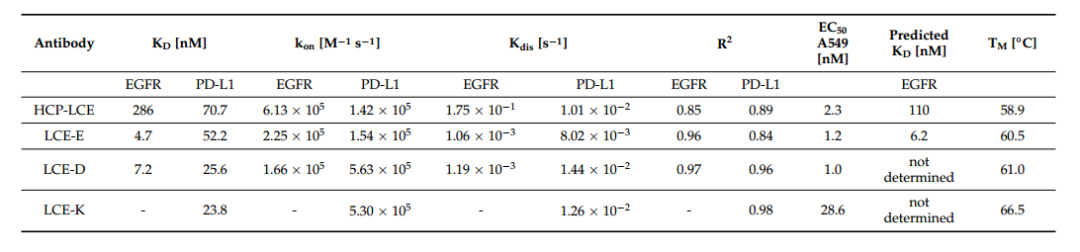
采用BLI测量确定 HCP- LCE 变体对EGFR和PD-L1的亲和力(图2)。突变体 LCE-D和LCE-E对EGFR的亲和力比野生型增加了约60倍,KD值在个位数纳摩尔范围(图2,表1),亲和力显著增加的主要原因是由于解离率提高,而LCE-K突变体不靶向EGFR,表明LCDR3第三位对 EGFR 结合影响较大。所有四种变体对PD-L1结合动力学相似,说明LCDR3突变对PD-L1结合无主要影响,PD-L1的KD值在两位数纳摩尔范围(表1)。
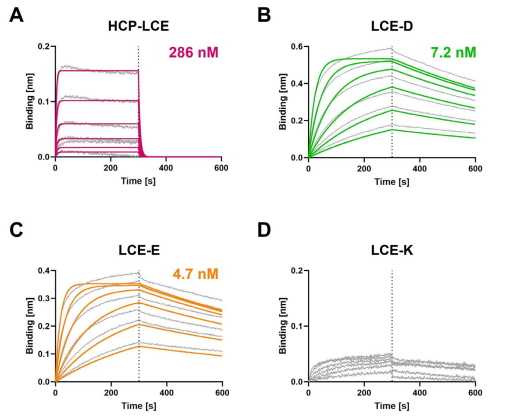
图2. 通过BLI测量表征HCP-LCE变体的EGFR结合。BLI测定(A) HCP-LCE、(B) LCE-D、(C) LCE-E和(D) LCE-K对EGFR的影响。LCE-D和LCE-E靶向EGFR的亲和力高于HCP-LCE,而突变体LCE-K不能结合EGFR。拟合用彩色曲线表示。
HCP-LCE和LCE-E与两种靶标的结合动力学在helix®生物传感器仪器上使用switchSENSE®技术进行了表征。DNA共轭靶标与荧光团标记的适配器链混合,后者混合在芯片表面上锚定链。分析物的结合是由荧光团感知的,荧光团对其局部环境的变化很敏感。在这种情况下,分析物结合导致了猝灭效应,可见为信号降低(图3A-D)。该技术还可以使用不同的荧光团标记不同的抗原,在双色实验中对同一表面上的抗原进行平行分析。此外,比较了两种抗体变体与EGFR和PD-L1的结合动力学,无论是在一个固定蛋白的表面上,还是在两个目标蛋白都固定的表面上。观察到的相互作用表现为两相解离,且解离速率有快有慢(图3)。在LCDR3 (LCE-E变体)中引入谷氨酸减少了快速解离速率的影响,并稳定了抗体与EGFR的结合(图3C)。
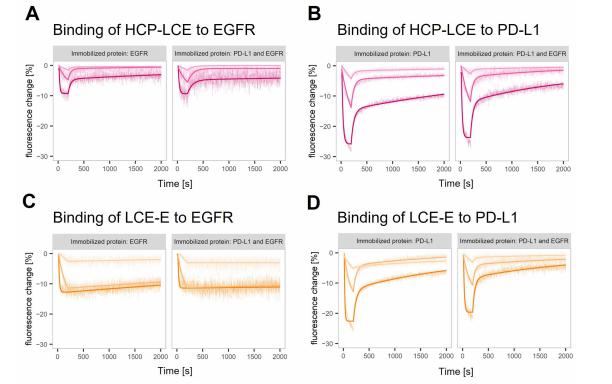
图3. 实时抗原结合测量单蛋白和双蛋白表面。(A,C) EGFR和(B,D) PD-L1结合(A,B) HCP-LCE和(C,D) LCE-E在EGFR或PD-L1表面和两种蛋白表面上的特性(每个图的左和右面板)。拟合模型假定所有观察到的相互作用均为双相解离。在LCE-E与EGFR的相互作用中,快速解离相的贡献最小。
为验证HCP-LCE突变体与肿瘤细胞亲和力,对EGFR和PD-L1双阳性A549细胞进行流式细胞仪结合实验。用不同浓度抗体染色细胞并检测结合情况。LCE-E和LCE-D显示特异性细胞结合,EC50值约为1nM,结合最大值相似(图4)。野生型HCP-LCE亲和力低,EC50为2.3nM且最大结合值显著降低。LCE-K因缺失EGFR结合特性,细胞结合也受影响(图4A,B)。
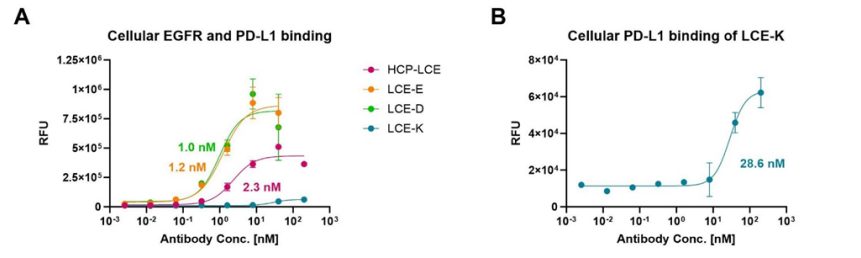
图4. HCP-LCE变体在EGFR/PD-L1双阳性A549细胞上的结合。(A) A549细胞上HCP-LCE(粉色)、LCE-E(橙色)、LCE-D(绿色)和LCE-K(蓝色)的细胞滴定。(B) A549细胞上LCE-K的细胞滴定;A所示图的放大视图。利用变斜率四参数拟合来拟合所得曲线。EC50值:HCP-LCE, 2.3 nM;LCE-E, 1.2 nM;LCE-D, 1.0 nM;LCE-K, 28.6 nM。所有的测量都是重复进行的,实验至少重复了三次,得到了相似的结果。
利用heliXcyto生物传感器设计原生分子互作细胞分析系统(RT-IC)实验,揭示改进的EGFR结合动力学对抗体变体LCE-E与靶阳性肿瘤细胞结合动力学的影响,并与野生型HCP-LCE进行比较。使用两种不同的EGFR和PDL1双阳性肿瘤细胞系A431和A549测量结合动力学。在A431细胞中,EGFR和PD-L1的表达水平高于A549细胞,且两种细胞系中EGFR的表达均高于PD-L1的表达[35]。将细胞装入heliXcyto生物传感器芯片上的细胞捕获网中,在增加抗体浓度的情况下,分三个注射步骤注射荧光标记的抗体变体。抗体的关联通过荧光信号的增加可见;切换到缓冲流后,信号的减少明显反映两者的解离。在A431细胞和A549细胞上检测到两种抗体(HCP-LCE和LCE-E)的结合(图5A-D)。
在大多数情况下,解离速率是双相的,并且以较慢的解离速率为主(图6)。以较慢的解离速率(KD2)计算的KD值在A431细胞上的HCP-LCE在0.1-0.7 nM范围内。HCP-LCE在A431上较低的KD2值是由于更快的结合速率引起的(图6A,B)。
在两种细胞系中,LCE-E显示较慢的第二次解离速率(kd2)(图6C,D)和较弱的第二次解离相对总解离振幅的贡献(图6C)。这导致亲和成熟抗体在细胞表面保留的时间增加(图6D)。
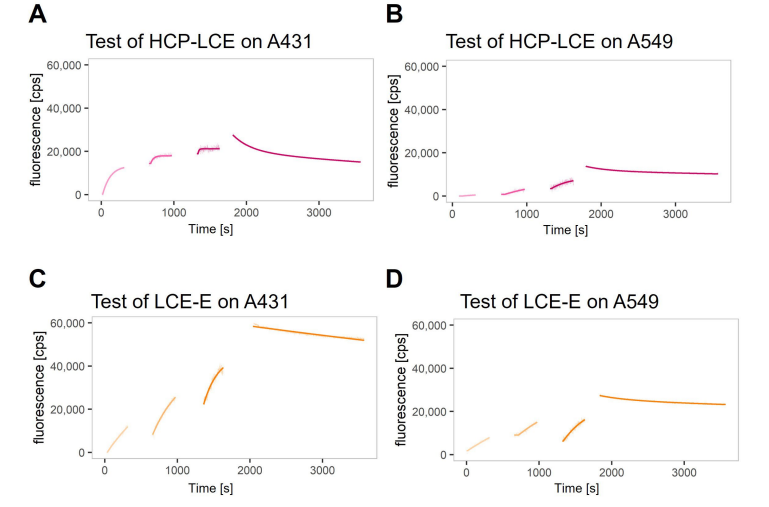
图5. 在EGFR和PD-L1双阳性的A431和A549细胞上的实时结合动力学。在helixcyto生物传感器中通过RT-IC测量的(A、B)HCP-LCE 和(C、D)LCE-E在(A、C)A431和(B、D)A549 细胞上的实时结合曲线。细胞用多聚甲醛固定,并加载到芯片上的五个独立细胞捕获网中。不断增加浓度并分三个连续注射步骤注入荧光标记的分析物。分析物的结合通过荧光信号的上升而明显显示出来。在第三次注射后,系统切换到缓冲液流以监测分析物的解离,表现为信号的明显降低。数据点用假设单相结合和双相解离的动力学模型进行拟合。
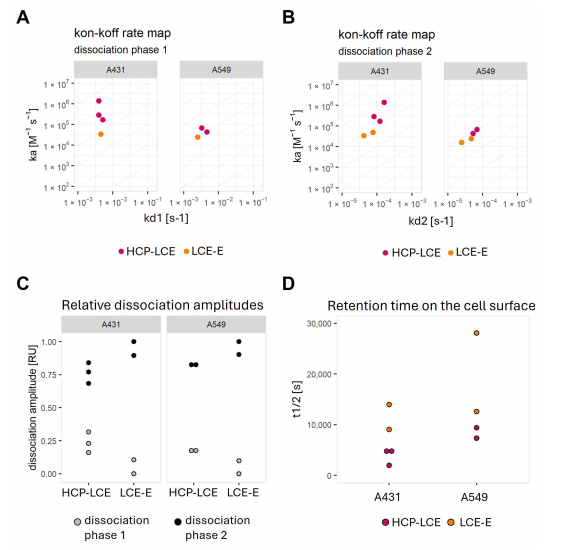
图6. 用假设分析物的单相结合、双相解离以及完全解离的动力学模型拟合并结合曲线后计算出的动力学值的图形。开-关速率图绘制(A)结合速率与解离速率1的关系,或(B)与解离速率2的关系。(C)展示较快解离速率(解离阶段 1)和较慢解离速率(解离阶段 2)相对贡献的图。(D)根据拟合模型为每种相互作用计算的半衰期值的图。
为研究LCE-E突变体与野生型HCP-LCE和其抗原的相互作用,使用AlphaFold multitimer[24,25]。结果显示,PD-L1结合主要通过重链CDRs(图7A),EGFR结合涉及重链和轻链CDRs(图7B),且HCDR3和LCDR3主要靶向EGFR表位,与实验数据一致[23]。PRODIGY web服务器预测蛋白-蛋白复合物的亲和力[32],结果显示 HCP-LCE与EGFR结合KD值为110 nM,LCE-E与EGFR结合KD值为6.2 nM,表明LCDR3氨基酸交换使亲和力显著增加,原因是LCDR3位置3的谷氨酸和EGFR 位置165的精氨酸形成盐桥所引起(图7C)。为了证明二合一VH:VL二聚体可以同时靶向EGFR和PD-L1,研究采用了蛋白对接方法[33,36]。PD-L1与EGFR:VH:VL复合物对接,形成一个四聚体蛋白复合物,其中两种抗原可以同时结合而没有空间位阻(图7D)。
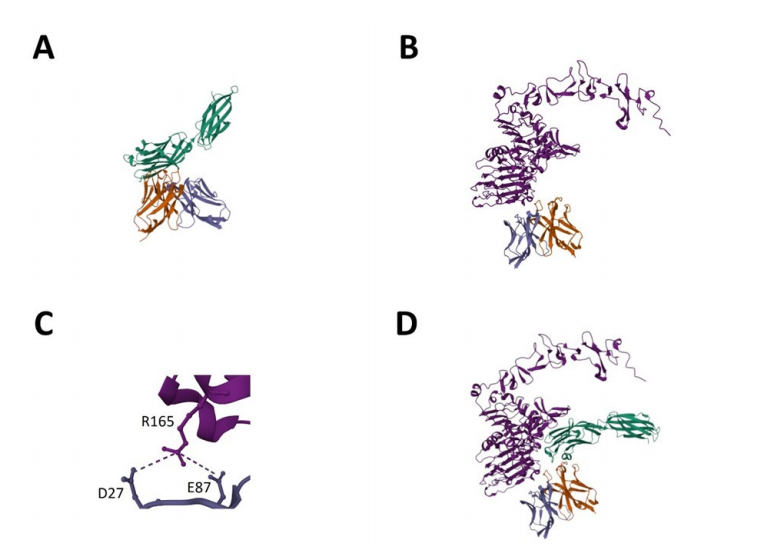
图7. 基于AlphaFold的LCE-E与EGFR和PD-L1结合的模型。(A)LCE-E与PD-L1 胞外结构域(绿色)的结合。(B)LCE-E与EGFR 胞外结构域(浅紫色)的结合。(C)LCE-E的LCDR3 处的谷氨酸(E87)与 EGFR胞外结构域 165 位的精氨酸(R165)之间的盐桥。(D)PD-L1与 EGFR:VH:VL 复合物对接的四聚体蛋白复合物。VH 片段显示为橙色,VL 片段显示为深紫色。使用AlphaFold Multimer创建。
亲和成熟是抗体获得增强亲和性和功能性的过程,由B细胞中免疫球蛋白基因体细胞超突变及抗原结合选择产生,通常发生在急性感染或接种疫苗后数周内[37]。与种系编码抗体相比,亲和成熟后的抗体高度突变且亲和力显著增加,是蛋白工程中提高体外亲和及结合作用、优化抗体治疗潜力的关键技术[38,39]。该研究生成鸡源性EGFR×PD-L1 二合一抗体亲和成熟变体以增强 EGFR 结合特性,通过位点饱和诱变,流式细胞仪检测随机YSD文库分离出变体LCE-E,其因LCDR3第三位氨基酸由Y变为E而改善了EGFR亲和力,AlphaFold模型表明可能是形成盐桥所致。
通过多种方法证实二合一变体LCE-E的EGFR结合亲和力提高,如BLI测量、混合表面实时抗原结合测量和肿瘤细胞结合实验等,其解离率降低使结合更稳定。但高亲和力不一定保证提高临床疗效,如帕利珠单抗变体虽效力提高了44倍,但体内疗效仅出现了适度改善,并且非特异性结合导致药代动力学谱较差[40]。HCP-LCE是嵌合抗体,由鸡源性VH和VL结构域移植到人IgG1支架上[23],鸡的重链和轻链产生与啮齿类动物不同,禽类V (D) J基因重排时只有小部分免疫球蛋白基因被选择,通过体细胞基因转化和超突变进一步多样化[41,42]。因HCP-LCE由两只鸡免疫产生,重链和轻链不在同一只鸡中,体内未亲和成熟,显示出体外亲和优化潜力。HCP-LCE同时靶向EGFR和PD-L1,抑制EGFR信号传导[23],也有其他研究描述抗原EGFR和PD-L1的有利组合。
总的来说,该研究已经提出了一种直接的方法来实现鸡源性二合一抗体的亲和力成熟,优化了两个靶点中只有一个与单个Fab片段同时处理的亲和力。对LCDR3区域的单个氨基酸进行突变,然后进行YSD文库生成和FACS筛选,结果分离出一种靶向EGFR的变异,其亲和力增加了60倍。BLI测量表明,亲和度的增加主要是由于解离率的提高。switchSENSE®与RT-IC的结果表明,LCE-E中LCDR3引入的谷氨酸降低了对PD- L1结合的快速解离率影响,对EGFR的结合更稳定且 LCE-E在A431和A549 细胞上的解离速率较HCP-LCE更慢,保留在细胞表面的时间更长。
Alphafold的模型预测,LCDR3中的谷氨酸与EGFR形成盐桥,导致亲和力增加。谷氨酸与赖氨酸的交换(LCE-K)完全消除了EGFR的结合特性。目前为止,这代表了第一个亲和成熟的二合一抗体,对其两个靶点之一的结合进行了优化。
该研究提出了一种鸡源Two-in-One抗体亲和力成熟的简单方法,优化了单个Fab 片段同时靶向的两个靶点中的一个靶点的亲和力,通过switchSENSE®与RT-IC等多种实验方法验证了变体特性。在抗体应用领域中,switchSENSE®可深入探究抗体与抗原的结合动力学,为理解抗体作用机制提供关键数据。RT-IC能够实时监测抗体在靶细胞上的结合情况,有助于评估抗体的靶向性和有效性。二者结合可为抗体的研发、优化和临床应用提供重要的技术支持和理论依据。

同腾睿杰(上海)生物科技有限公司作为Dynamic Biosensors中国总代理商,为您提供优质的售前售后服务。
联系电话:021-50826962
联系邮箱:sales@ttbiotech.com
[1]. Hansel, T.T.; Kropshofer, H.; Singer, T.; Mitchell, J.A.; George, A.J.T. The safety and side effects of monoclonal antibodies. Nat. Rev.Drug Discov. 2010, 9, 325–338.
[2]. Stanfield, R.L.; Wilson, I.A. Antibody Structure. Microbiol. Spectr. 2014, 2.
[3]. Tsuji, I.; Vang, F.; Dominguez, D.; Karwal, L.; Sanjali, A.; Livengood, J.A.; Davidson, E.; Fouch, M.E.; Doranz, B.J.; Das, S.C.;et al. Somatic Hypermutation and Framework Mutations of Variable Region Contribute to Anti-Zika Virus-Specific Monoclonal Antibody Binding and Function. J. Virol. 2022, 96, e0007122.
[4]. Green, N.S.; Lin, M.M.; Scharff, M.D. Somatic hypermutation of antibody genes: A hot spot warms up. Bioessays 1998, 20, 227–234.
[5]. Tas, J.M.J.; Mesin, L.; Pasqual, G.; Targ, S.; Jacobsen, J.T.; Mano, Y.M.; Chen, C.S.; Weill, J.-C.; Reynaud, C.-A.; Browne, E.P.; et al. Visualizing antibody affinity maturation in germinal centers. Science 2016, 351, 1048–1054.
[6]. Strohl, W.R.; Strohl, L.M. Sources of antibody variable chains. In Therapeutic Antibody Engineering; Elsevier: Amsterdam, The Netherlands, 2012; pp. 77–595, ISBN 9781907568374.
[7]. Bogen, J.P.; Elter, A.; Grzeschik, J.; Hock, B.; Kolmar, H. Humanization of Chicken-Derived Antibodies by Yeast Surface Display. Methods Mol. Biol. 2022, 2491, 335–360.
[8]. Tabasinezhad, M.; Talebkhan, Y.; Wenzel, W.; Rahimi, H.; Omidinia, E.; Mahboudi, F. Trends in therapeutic antibody affinity maturation: From in-vitro towards next-generation sequencing approaches. Immunol. Lett. 2019, 212, 106–113.
[9]. Persson, H.; Kirik, U.; Thörnqvist, L.; Greiff, L.; Levander, F.; Ohlin, M. In Vitro Evolution of Antibodies Inspired by In Vivo Evolution. Front. Immunol. 2018, 9, 1391.
[10]. Lou, J.; Marks, J.D. Affinity Maturation by Chain Shuffling and Site Directed Mutagenesis. In Antibody Engineering; Kontermann, R., Dübel, S., Eds.; Scholars Portal: Berlin/Heidelberg, Germany, 2010; pp. 377–396, ISBN 978-3-642-01143-6.
[11]. Smith, G.P. Filamentous fusion phage: Novel expression vectors that display cloned antigens on the virion surface. Science 1985, 228, 1315–1317.
[12]. Boder, E.T.; Wittrup, K.D. Yeast surface display for screening combinatorial polypeptide libraries. Nat. Biotechnol. 1997, 15, 553–557.
[13]. Tsuruta, L.R.; Dos, M.L.; Moro, A.M. Display Technologies for the Selection of Monoclonal Antibodies for Clinical Use. In Antibody Engineering; Böldicke, T., Ed.; InTech: London, UK, 2018; ISBN 978-953-51-3825-9.
[14]. He, M.; Khan, F. Ribosome display: Next-generation display technologies for production of antibodies in vitro. Expert Rev. Proteomics 2005, 2, 421–430.
[15]. Chowdhury, P.S.; Pastan, I. Improving antibody affinity by mimicking somatic hypermutation in vitro. Nat. Biotechnol. 1999, 17, 568–572.
[16]. Rajpal, A.; Beyaz, N.; Haber, L.; Cappuccilli, G.; Yee, H.; Bhatt, R.R.; Takeuchi, T.; Lerner, R.A.; Crea, R. A general method for greatly improving the affinity of antibodies by using combinatorial libraries. Proc. Natl. Acad. Sci. USA 2005, 102, 8466–8471.
[17]. Laffly, E.; Pelat, T.; Cédrone, F.; Blésa, S.; Bedouelle, H.; Thullier, P. Improvement of an antibody neutralizing the anthrax toxin by simultaneous mutagenesis of its six hypervariable loops. J. Mol. Biol. 2008, 378, 1094–1103.
[18]. Chen, S.; Li, J.; Li, Q.; Wang, Z. Bispecific antibodies in cancer immunotherapy. Hum. Vaccin. Immunother. 2016, 12, 2491–2500.
[19]. Sheridan, C. Bispecific antibodies poised to deliver wave of cancer therapies. Nat. Biotechnol. 2021, 39, 251–254.
[20]. Krishnamurthy, A.; Jimeno, A. Bispecific antibodies for cancer therapy: A review. Pharmacol. Ther. 2018, 185, 122–134.
[21]. Ju, X.; Zhang, H.; Zhou, Z.; Wang, Q. Regulation of PD-L1 expression in cancer and clinical implications in immunotherapy. Am. J. Cancer Res. 2020, 10, 1–11.
[22]. Sigismund, S.; Avanzato, D.; Lanzetti, L. Emerging functions of the EGFR in cancer. Mol. Oncol. 2018, 12, 3–20.
[23]. Harwardt, J.; Bogen, J.P.; Carrara, S.C.; Ulitzka, M.; Grzeschik, J.; Hock, B.; Kolmar, H. A Generic Strategy to Generate Bifunctional Two-in-One Antibodies by Chicken Immunization. Front. Immunol. 2022, 13, 888838.
[24]. Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589.
[25]. Mirdita, M.; Schütze, K.; Moriwaki, Y.; Heo, L.; Ovchinnikov, S.; Steinegger, M. ColabFold: Making protein folding accessible to all. Nat. Methods 2022, 19, 679–682.
[26]. Baek, M.; DiMaio, F.; Anishchenko, I.; Dauparas, J.; Ovchinnikov, S.; Lee, G.R.; Wang, J.; Cong, Q.; Kinch, L.N.; Schaeffer, R.D.; et al. Accurate prediction of protein structures and interactions using a three-track neural network. Science 2021, 373, 871–876.
[27]. Evans, R.; O’Neill, M.; Pritzel, A.; Antropova, N.; Senior, A.; Green, T.; Žídek, A.; Bates, R.; Blackwell, S.; Yim, J.; et al. Protein complex prediction with AlphaFold-Multimer. bioRxiv 2021.
[28]. Mirdita, M.; Steinegger, M.; Söding, J. MMseqs2 desktop and local web server app for fast, interactive sequence searches. Bioinformatics 2019, 35, 2856–2858.
[29]. Steinegger, M.; Söding, J. MMseqs2 enables sensitive protein sequence searching for the analysis of massive data sets. Nat. Biotechnol. 2017, 35, 1026–1028.
[30]. Suzek, B.E.; Wang, Y.; Huang, H.; McGarvey, P.B.; Wu, C.H. UniRef clusters: A comprehensive and scalable alternative for improving sequence similarity searches. Bioinformatics 2015, 31, 926–932.
[31]. Eastman, P.; Swails, J.; Chodera, J.D.; McGibbon, R.T.; Zhao, Y.; Beauchamp, K.A.; Wang, L.-P.; Simmonett, A.C.; Harrigan, M.P.; Stern, C.D.; et al. OpenMM 7: Rapid development of high performance algorithms for molecular dynamics. PLoS Comput. Biol. 2017, 13, e1005659.
[32]. Vangone, A.; Bonvin, A.M.J.J. PRODIGY: A Contact-based Predictor of Binding Affinity in Protein-protein Complexes. Bio Protoc. 2017, 7, e2124.
[33]. Yan, Y.; Tao, H.; He, J.; Huang, S.-Y. The HDOCK server for integrated protein-protein docking. Nat. Protoc. 2020, 15, 1829–1852.
[34]. Siloto, R.M.; Weselake, R.J. Site saturation mutagenesis: Methods and applications in protein engineering. Biocatal. Agric. Biotechnol. 2012, 1, 181–189.
[35]. Bogen, J.P.; Carrara, S.C.; Fiebig, D.; Grzeschik, J.; Hock, B.; Kolmar, H. Design of a Trispecific Checkpoint Inhibitor and Natural Killer Cell Engager Based on a 2 + 1 Common Light Chain Antibody Architecture. Front. Immunol. 2021, 12, 669496.
[36]. Yin, R.; Feng, B.Y.; Varshney, A.; Pierce, B.G. Benchmarking AlphaFold for protein complex modeling reveals accuracy determinants. Protein Sci. 2022, 31, e4379.
[37]. MacLennan, I.C. Germinal centers. Annu. Rev. Immunol. 1994, 12, 117–139.
[38]. Chan, T.D.; Brink, R. Affinity-based selection and the germinal center response. Immunol. Rev. 2012, 247, 11–23.
[39]. Chan, D.T.Y.; Groves, M.A.T. Affinity maturation: Highlights in the application of in vitro strategies for the directed evolution of antibodies. Emerg. Top. Life Sci. 2021, 5, 601–608.
[40]. Sassi, A.B.; Nagarkar, R.; Hamblin, P. Biobetter Biologics. Novel Approaches and Strategies for Biologics, Vaccines and Cancer Therapies; Elsevier: Amsterdam, The Netherlands, 2015; pp. 199–217, ISBN 9780124166035.
[41]. Kurosawa, K.; Ohta, K. Genetic diversification by somatic gene conversion. Genes 2011, 2, 48–58.
[42]. Mallaby, J.; Mwangi, W.; Ng, J.; Stewart, A.; Dorey-Robinson, D.; Kipling, D.; Hershberg, U.; Fraternali, F.; Nair, V.; Dunn-Walters, D. Diversification of immunoglobulin genes by gene conversion in the domestic chicken (Gallus gallus domesticus). Discov. Immunol. 2023, 2, kyad002.

