
由于绝大多数细菌和古细菌尚未被成功培养,因此它们的基因组、新陈代谢潜力以及在环境中的功能仍未得到充分研究;我们将这些微生物称为微生物暗物质[1-5]。单细胞基因组学是研究微生物暗物质的重要手段,但全基因组扩增步骤存在诸多挑战。传统全基因组扩增方法(MDA)虽常用,但成本高且对特定基因组区域存在扩增偏差,导致基因组覆盖不均,限制了其在高通量研究中的应用,难以获取高质量基因组,尤其是微生物群落中的少数类群基因组[6,7]。
在微生物研究领域,单细胞基因组学(SCG)为探索微生物暗物质(MDM)提供了关键途径[8-10]。然而,全基因组扩增这一关键步骤面临诸多挑战,如成本高昂、扩增偏差以及污染问题等,严重制约了研究的深入开展。
I.DOT非接触式移液器的技术核心及优势

非接触式液体转移:在纳升至微升范围内,实现极低死体积(<1 μL)和零交叉污染风险的液体转移,为细胞培养提供前所未有的精确度和安全性。
Drop Detection系统:对每个液滴进行计数和检测,确保实验的可重复性和可靠性,提高实验效率。
本篇推文主要介绍I.DOT 技术在微生物单细胞全基因组扩增研究中的应用。旨在解决传统全基因组扩增方法(如MDA)成本高、扩增偏差大及污染等问题,为微生物单细胞基因组学研究提供新方法。
实验方法
第一步:细胞培养与分离
培养大肠杆菌 K12 MG1655 至指数生长期,在UV-去污染的 ISO 4级洁净室中处理,用 BD FACS Melody 将细胞分选至384孔板,低温保存。
第二步:细胞裂解
采用改良裂解缓冲液[11],通过I.DOT mini将裂解液分配到细胞上并放入不含细胞的孔中(阴性对照)。在21℃下孵育10分钟,通过加入等体积的中和缓冲液来中和。
第三步:多重置换扩增(MDA)
用 REPLI-g单细胞试剂盒进行MDA反应,然后用I.DOT mini将REPLI-g主混合液分配到裂解的细胞和阴性对照上,使最终的MDA体积为0.5, 0.8, 1.0, 1.25, 5, 10 µL。MDA在CFX-384热循环器(Bio-Rad, Hercules,CA,USA)中在30℃下孵育6小时,然后在65◦C下孵育10分钟以停止扩增并保持在4℃。扩增的DNA保存在−20℃,直到用于文库准备。
第四步:文库制备与测序
在紫外线净化的层流PCR工作台进行相关操作,包括纯化、文库制备,最后用 Illumina NextSeq 550 测序。
第五步:数据处理及分析
使用多种工具进行质量检测、修剪、归一化、去污染、去重、比对、组装和质量评价[7,12-19]
1、使用FastQC和Trim Galore软件对序列数据进行质量检查和修剪。
2、用BBTools软件将修剪后的数据标准化到约200倍的测序深度,并进行了不同深度(100×, 80×, 60×, 40×, 20×)的下采样,以分析测序深度对覆盖度的影响。
3、使用FASTQ-Screen软件对数据进行污染评估,并保留大肠杆菌的多映射读取。
4、利用BBTools软件去除PCR重复序列。
5、将读取数据映射到大肠杆菌MG1655参考基因组,并计算10 kb区间的覆盖度。
6、在进行de novo组装前,使用bbnorm.sh软件对读取覆盖度进行标准化。
7、使用SPAdes软件进行单细胞模式下的组装,并用QUAST和MDMcleaner软件评估组装质量和污染程度。
8、使用Microsoft Excel进行统计分析,包括ANOVA和Kruskal-Wallis测试,以及t-Test进行成对比较。
9、使用R软件的ineq包计算Gini指数,并用ggplot2创建读取深度和Lorenz曲线图。
实验数据分析
1、在384孔板上进行总反应体积为0.5、0.8、1.0、1.25、5.0和10 µL的MDAs(表A1)。最小浓度0.5µL的MDA反应不起作用,0.8 µL和1.0 µL反应的扩增成功率分别只有68.75%和62.50%。相比之下,1.25 µL MDA反应的成功率为87.50%,而5.0 µL和10 µL MDA反应的成功率均为100%。在较低的丙二醛反应体系中,较低的成功率可能是由于在小体积中聚合酶的蒸发或位阻[20,21]。平均而言,放大达到检测阈值所需的时间(用Cq);1.25 µL的MDA反应体积(图1A)反应时间最早。先前的研究报道,Cq值越早,基因组恢复成功率和质量越高[11,22]。此外,我们检测到的相对荧光(RFU)终点和成功反应的DNA产率随着反应体积的减少而下降(图1A,B),最初表明体积的减少可能限制了MDA的指数性质[23,24],这应该提高基因组的覆盖率和均匀性。为了进一步比较WGA反应的质量,根据Cq和RFU值选择每种不同MDA反应体积的5个扩增重复,然后使用等量的DNA进行Illumina测序。
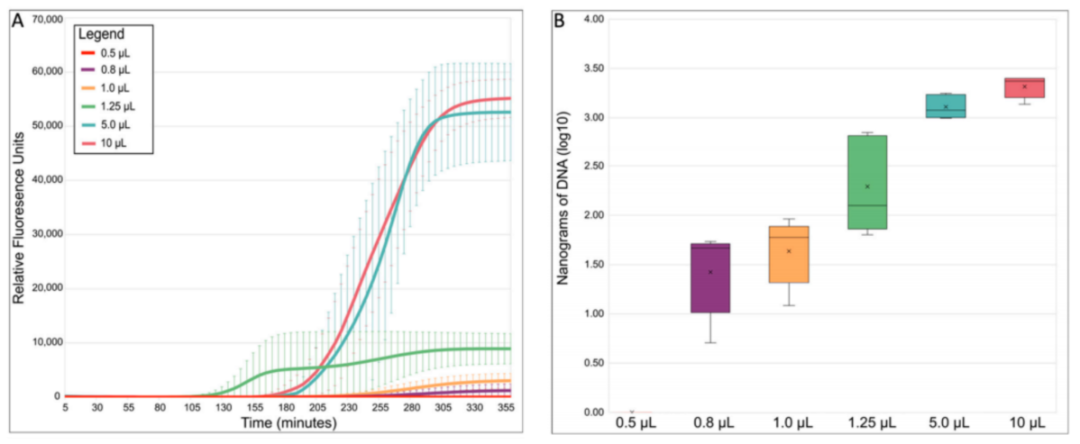
图1. MDA反应统计概述。(A)平均丙二醛反应动力学的反应大小。标准误差条表示使用每个反应体积的所有五个重复计算的标准偏差。(B)按反应大小计算的MDA平均扩增率。相对荧光单(RFU)指SYTO™-13用实时热循环仪测得的荧光信号。SYTO™-13用于监测MDA的进展,因为它在扩增时与双链DNA结合。方框的中线代表中位数,x代表平均值。用5个重复进行计算。
2、不同MDA反应体积在读取修剪过程中丢失的读取数量存在显著差异(图2A),其中1.25 µL的MDA反应体积在读取质量修剪中丢失的读取数量显著少于其他体积(p ≤ 0.05)。修剪后,所有样本被标准化到200×测序深度,以便公平比较不同反应体积的映射和组装质量。尽管较大体积反应的平均重复读取数量较多(图2B),但差异并不显著(p = 0.0870)。这可能与大体积MDA反应中模板特异性较低有关[25,26],导致更多的错误引物结合和扩增,尤其是在模板浓度非常低时。此外,较大的反应体积在过滤后去除的污染读取也较多(图2C),这可能是由于背景污染与大肠杆菌单细胞DNA之间的竞争增加所致[25,27]。总的来说,5和10 µL的MDA反应体积结果一致性较差,表现为重复之间的变异性较大(图2)。
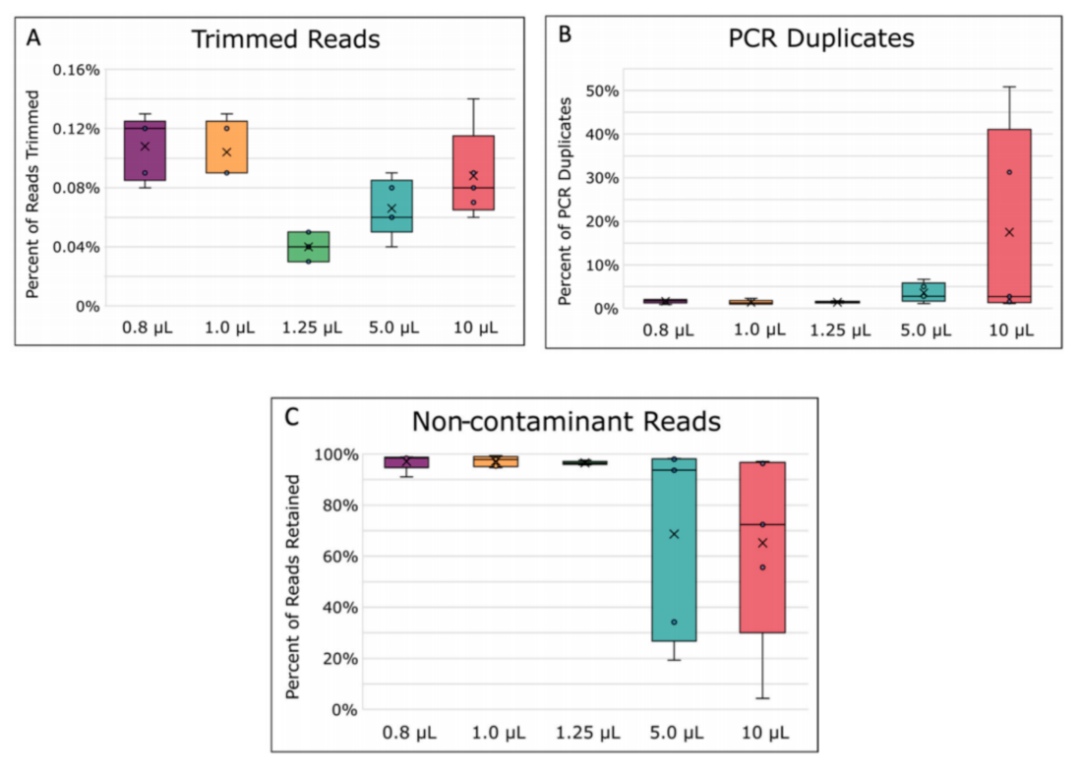
图2. 读取处理统计数据。(A)在质量修剪期间去除读数的百分比。(B)去除PCR重复的百分比。(C)读数污染物过滤后保留的读数百分比。方框的中线代表中位数,x代表平均值。用5个重复进行计算
3、由于模板特异性较低,高于1.25 µL的MDA反应量在对参比大肠杆菌MG1655基因组的读取定位中也表现较差,如基因组覆盖宽度和覆盖均匀性所示(图3A,B)。而1.25 µL的反应体积在384孔板中被认为是改善MDA的“最佳点”。1.25 µL MDA反应体积的平均读取覆盖了大肠杆菌基因组的85%,比其他体积的反应高出19%到40%(图3A)。与WGA-X™方法相比,我们的覆盖度提高了约19%,即使在读取映射过程中使用了大约200万更少的读取。此外,1.25 µL反应体积的覆盖度均匀性更高,这通过Lorenz曲线和Gini指数(衡量覆盖度均匀性的指标)得到了证实,Gini指数在1.25 µL反应中最低,表明其覆盖度最均匀(图5B)。这些结果表明,1.25 µL的MDA反应体积在提高基因组覆盖度和均匀性方面具有显著优势。
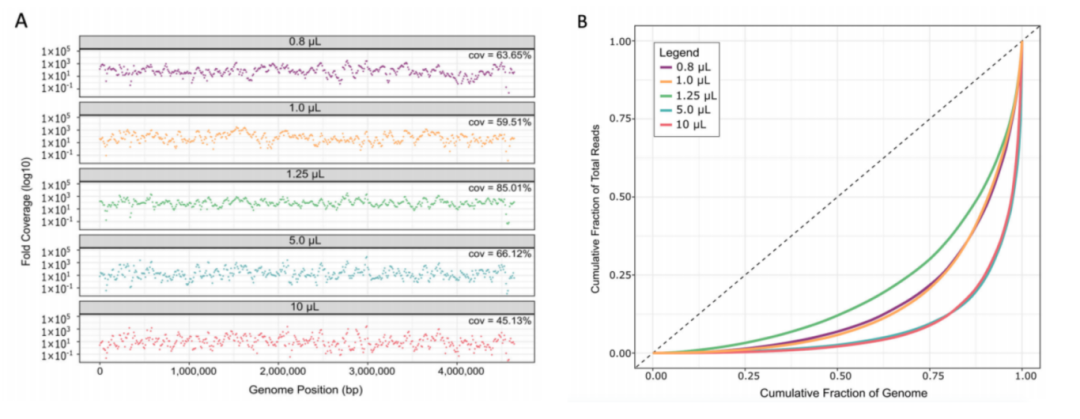
图3. 基因组覆盖率和覆盖一致性偏差。(A) 在大肠杆菌基因组的10kb区间内计算每个复制的读取深度。图表显示了每个反应体积的所有重复的平均值。Cov. 是平均覆盖宽度,即至少一次读取覆盖的基因组位置的百分比。(B)在大肠杆菌基因组的10kb区间内计算读取覆盖率和深度的均匀性,并对每个反应体积的所有五个重复进行平均。
4、对所有复制的单放大基因组(SAGs)进行了组装和比较。结果显示,由于MDA引入的大差异,我们在组装前将读取深度标准化至100×的目标深度。然而,小于和大于1.25 µL的MDA反应体积由于在预处理步骤中丢失了更多读取,导致最终序列深度较低,从而使得组装质量较1.25 µL MDA反应体积的组装结果差(图4A-C)。具体来说,1.25 µL反应的平均总长度和N50(代表50%总序列长度的最短contig序列长度)分别为3,522,851 bp和46,179 bp(图4B,C),表明这些组装更加连续,质量更高。同时计算了组装覆盖度和完整性,覆盖度是指与参考基因组映射的组装(contigs)的百分比[70],而基因组完整性则是通过MDMcleaner估计标记基因的存在。1.25 µL MDA反应的组装覆盖度和完整性显著高于其他体积,平均约为75%和94%,污染最低(图6D-F)。其中三个1.25 µL MDA反应复制的覆盖度甚至超过了75%,最高达到89.5%。相比之下,WGA-X™即使使用了约5倍多的读取,大肠杆菌组装覆盖度也低于60%。我们的10 µL MDA反应的组装覆盖度与WGA-X™在10 µL反应中报告的范围一致,这表明WGA-X™也可能从进一步的体积减少中受益。与其他体积减少方法相比,我们的更高组装覆盖度与之前报告的大肠杆菌MDA在皮升级液滴(88-91%)[28]和纳升级液滴(88-94%)[27]中的覆盖度范围一致。
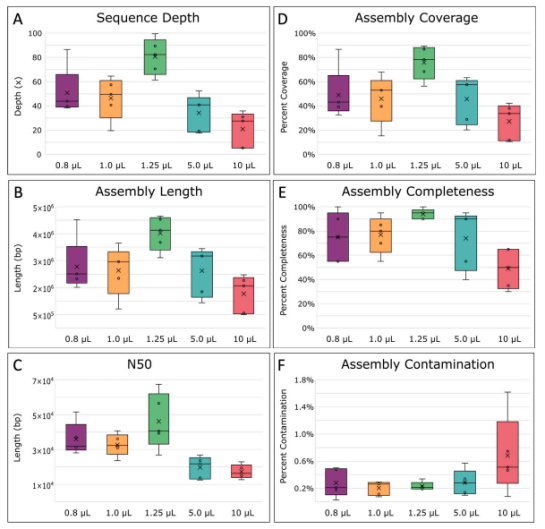
图4. 单扩增基因组(SAG)组装统计。(A)最终序列深度,以基因组内每个碱基平均测序的估计次数计算。(B)组合的总平均长度,(C) N50平均值,支持50%基因组组合所需的最小组合长度,(D)整个基因组中组合的覆盖率百分比。大肠杆菌MG1655参考基因组,均采用QUAST测定[13]。(E)组装基因组的完整性和(F)组装中受污染碱基的百分比,用MDMCleaner[7]测定。方框的中线代表中位数,x代表平均值。用5个重复进行计算。
总结与讨论
总体而言,1.25 μL 的 MDA 反应体积显著优于传统 50 μL反应,成本降低约 97.5%,基因组覆盖度和均匀性大幅提升,产生更完整、偏差更小的单扩增基因组(SAGs)。相比其他体积降低方法,该方法在标准384孔板中更易操作,可提高微生物单细胞基因组学研究可行性,有助于深入探索微生物多样性和功能,特别是针对稀有物种和微生物暗物质研究。I.DOT技术在本研究中的应用,不仅提高了单细胞基因组扩增的效率和准确性,还降低了成本,使得SCG更易于在未来的研究中应用。这项技术为研究3D细胞培养模型中的细胞信号传导提供了强大工具,有助于在更接近生理条件下研究各种生物学过程,为组织工程和再生医学的发展提供新的思路和方法。
同腾睿杰(上海)生物技有限公司作为CYTENA I.DOT中国总代理商,为您提供优质的售前售后服务。
联系电话:021-50826962
联系邮箱:sales@ttbiotech.com
参考文献
1. Wu, D.; Raymond, J.; Wu, M.; Chatterji, S.; Ren, Q.; Graham, J.E.; Bryant, D.A.; Robb, F.; Colman, A.; Tallon, L.J.; et al. Complete Genome Sequence of the Aerobic CO-Oxidizing Thermophile Thermomicrobium Roseum. PLoS ONE 2009, 4, e4207.
2. McDonald, D.; Price, M.N.; Goodrich, J.; Nawrocki, E.P.; Desantis, T.Z.; Probst, A.; Andersen, G.L.; Knight, R.; Hugenholtz, P. An Improved Greengenes Taxonomy with Explicit Ranks for Ecological and Evolutionary Analyses of Bacteria and Archaea. ISME J. 2012, 6, 610–618.
3. Hug, L.A.; Baker, B.J.; Anantharaman, K.; Brown, C.T.; Probst, A.J.; Castelle, C.J.; Butterfield, C.N.; Hernsdorf, A.W.; Amano, Y.; Ise, K. A New View of the Tree of Life. Nat. Microbiol. 2016, 1, 16048.
4. Lloyd, K.G.; Steen, A.D.; Ladau, J.; Yin, J.; Crosby, L. Phylogenetically Novel Uncultured Microbial Cells Dominate Earth Microbiomes. mSystems 2018, 3, e00055-18.
5. Solden, L.; Lloyd, K.; Wrighton, K. The Bright Side of Microbial Dark Matter: Lessons Learned from the Uncultivated Majority. Curr. Opin. Microbiol. 2016, 31, 217–226
6. Dick, G.J.; Andersson, A.F.; Baker, B.J.; Simmons, S.L.; Thomas, B.C.; Yelton, A.P.; Banfield, J.F. Community-Wide Analysis of Microbial Genome Sequence Signatures. Genome Biol. 2009, 10, R85.
7. Vollmers, J.; Wiegand, S.; Lenk, F.; Kaster, A.-K. How Clear Is Our Current View on Microbial Dark Matter? (Re-)Assessing Public MAG & SAG Datasets with MDMcleaner. Nucleic Acids Res. 2022, 50, e76
8. Kaster, A.K.; Sobol, M.S. Microbial Single-Cell Omics: The Crux of the Matter. Appl. Microbiol. Biotechnol. 2020, 104, 8209–8220.
9. Rinke, C.; Lee, J.; Nath, N.; Goudeau, D.; Thompson, B.; Poulton, N.; Dmitrieff, E.; Malmstrom, R.; Stepanauskas, R.; Woyke, T. Obtaining Genomes from Uncultivated Environmental Microorganisms Using FACS-Based Single-Cell Genomics. Nat. Protoc. 2014, 9, 1038–1048.
10. Stepanauskas, R. Single Cell Genomics: An Individual Look at Microbes. Curr. Opin. Microbiol. 2012, 15, 613–620.
Stepanauskas, R.; Fergusson, E.A.; Brown, J.; Poulton, N.J.; Tupper, B.; Labonté, J.M.;
11. Becraft, E.D.; Brown, J.M.; Pachiadaki, M.G.; Povilaitis, T.; et al. Improved Genome Recovery and Integrated Cell-Size Analyses of Individual Uncultured Microbial Cells and Viral Particles. Nat. Commun. 2017, 8, 84.
12. Zeileis, A.; Kleiber, C.; Rep, M.A.Z.-T. 2009, U. Package “Ineq.”. 2014. Available online: https://cran.microsoft.com (accessed on1 August 2022)
13. Mikheenko, A.; Prjibelski, A.; Saveliev, V.; Antipov, D.; Gurevich, A. Versatile Genome Assembly Evaluation with QUAST-LG. Bioinformatics 2018, 34, i142–i150.
14. Krueger, F.; James, F.; Ewels, P.; Afyounian, E.; Schuster-Boeckler, B. Trim Galore. Available online: https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/ (accessed on 4 September 2020).
15.Bushnell, B. BBtools Software Package. Available online: https://sourceforge.net/projects/bbmap/ (accessed on 9 October 2020).
16. Wingett, S.W.; Andrews, S. FastQ Screen: A Tool for Multi-Genome Mapping and Quality Control. F1000Research 2018, 7, 1338.
17. Prjibelski, A.; Antipov, D.; Meleshko, D.; Lapidus, A.; Korobeynikov, A. Using SPAdes De Novo Assembler. Curr. Protoc. Bioinform. 2020, 70, e102.
18. R Core Team. R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing; Vienna, Austria. 2020. Available online: https://www.R-project.org/ (accessed on 29 February 2020).
19. Villanueva, R.A.M.; Chen, Z.J. Ggplot2: Elegant Graphics for Data Analysis (2nd Ed.). Measurement 2019, 17, 160–167.
20. Kuznetsova, I.M.; Turoverov, K.K.; Uversky, V.N. What Macromolecular Crowding Can Do to a Protein. Int. J. Mol. Sci. 2014, 15, 23090–23140.
21. Ralston, G.B. Effects of Crowding in Protein Solutions. J. Chem. Educ. 1990, 67, 857–860.
22. Labonté, J.M.; Field, E.K.; Lau, M.; Chivian, D.; Van Heerden, E.; Wommack, K.E.; Kieft, T.L.; Onstott, T.C.; Stepanauskas, R. Single Cell Genomics Indicates Horizontal Gene Transfer and Viral Infections in a Deep Subsurface Firmicutes Population. Front.Microbiol. 2015, 6, 349.
23. Lasken, R.S. Single-Cell Sequencing in Its Prime. Nat. Biotechnol. 2013, 31, 211–212.
24. Rhee, M.; Light, Y.K.; Meagher, R.J.; Singh, A.K. Digital Droplet Multiple Displacement Amplification (ddMDA) for Whole Genome Sequencing of Limited DNA Samples. PLoS ONE 2016, 11, e0153699.
25. Marcy, Y.; Ishoey, T.; Lasken, R.S.; Stockwell, T.B.; Walenz, B.P.; Halpern, A.L.; Beeson, K.Y.; Goldberg, S.M.D.; Quake, S.R.Nanoliter Reactors Improve Multiple Displacement Amplification of Genomes from Single Cells. PLoS Genet. 2007, 3, 1702–1708.
26. Leung, K.; Klaus, A.; Lin, B.K.; Laks, E.; Biele, J.; Lai, D.; Bashashati, A.; Huang, Y.-F.F.; Aniba, R.; Moksa, M.; et al. Robust High-Performance Nanoliter-Volume Single-Cell Multiple Displacement Amplification on Planar Substrates. Proc. Natl. Acad. Sci. USA 2016, 113, 8484–8489.
27. Gole, J.; Gore, A.; Richards, A.; Chiu, Y.J.; Fung, H.L.; Bushman, D.; Chiang, H.I.; Chun, J.; Lo, Y.H.; Zhang, K. Massively Parallel Polymerase Cloning and Genome Sequencing of Single Cells Using Nanoliter Microwells. Nat. Biotechnol. 2013, 31, 1126–1132.
28. Nishikawa, Y.; Hosokawa, M.; Maruyama, T.; Yamagishi, K.; Mori, T.; Takeyama, H. Monodisperse Picoliter Droplets for Low-Bias and Contamination-Free Reactions in Single-Cell Whole Genome Amplification. PLoS ONE 2015, 10, e0138733.

