
在组织工程和再生医学领域,人工多细胞系统正变得日益重要。然而,体外重建复杂的组织结构充满挑战,急需能够可控且高通量制造复杂多细胞结构的方法。该文介绍一种创新的3D细胞培养方法——基于可编程液滴融合技术的多球状细胞结构组装(proMAD方法)。这一方法不仅能够精确控制细胞组成和空间分布,还能在迷你化、高通量的格式下重建模拟天然组织的复杂性,可应用于细胞信号、肿瘤侵袭、胚胎发生和神经发育等多种生物过程的研究。
I.DOT非接触式移液器:
proMAD方法的技术核心及优势

proMAD方法无需支架,可以以高通量和小型化的方式自动形成球形组件阵列,以解决细胞-细胞通信中使用的细胞机制的复杂性。该方法的核心在于I.DOT非接触式移液器,它能够在纳升至微升的范围内,非接触式地将液体从源头转移到目标板。这意味着极低的死体积(<1 μL)和零交叉污染风险,为细胞培养提供了前所未有的精确度和安全性。
与此同时,I.DOT搭载的DropDetection系统,能够对每个液滴进行计数和检测,提供液体转移性能和总体积的即时反馈。这不仅确保了实验的可重复性和可靠性,还极大地提高了实验效率。
利用该文作者已开发的液滴微阵列(DMA)平台[1-3],它可以制造纳米升液滴微阵列,其中液滴的形状、大小和密度取决于被超疏水屏障包围的亲水性图案的设计。DMA平台可以在数百个单独的纳升液滴中培养和高通量筛选各种细胞类型,这些液滴作为独立的小型化栖息地[4]。
在DMA平台上使用悬滴法制备单细胞球体,使用了由14 × 14个亲水点(每个1 mm × 1 mm)组成的DMA,点之间的距离为500 µm(图1)。以HepG2细胞为例,将细胞悬液以200 nL的液滴形式通过I.DOT非接触式移液器分配在DMA上,每个液滴含有的细胞数量可控制球体直径(30-150 μm)。然后将DMA倒置,使细胞在液滴-空气界面因重力驱动聚集,培养2天后形成单个球体(图1a,1b)。采用以下打印顺序:为融合4个液滴,将培养基依次添加到D2、E2、D3、E3点(图1c中蓝色点,每个点多添加900 nL DMEM);为了融合3个液滴,将培养基依次添加到A1、A2、B2点(图1f中蓝色点,每个点增加850 nL DMEM)。然后相邻的液滴融合成一个。将载玻片倒置培养24、48、72、96 h。用Keyence软件对球体或合并球体进行观察和成像。
proMAD方法的基础是将额外体积的细胞培养基分配到相邻的单个液滴中,并控制它们在超疏水屏障上的融合(图1)。在相邻的每个200 nL的液滴中加入900 nL的介质(即每个点总共加入1100 nL),相邻的液滴会自发合并成一个新的大液滴,并牢固地附着在两个亲水点上(图1)。合并液滴后,将DMA载玻片再次倒置进行培养,使先前分离的两个球体在液滴底部接触,并使它们粘附融合成多球体复合物(图1a)。同样,通过增加多个相邻液滴的体积来合并两个以上的液滴(图1c-h)。有趣的是,这导致了由多个单独的细胞球体组成的融合球体的形成。为了融合两种不同的细胞系,选择了两种不同的细胞系:HeLa RFP细胞和HEK 293T细胞。用共聚焦显微镜检查合并的球体。
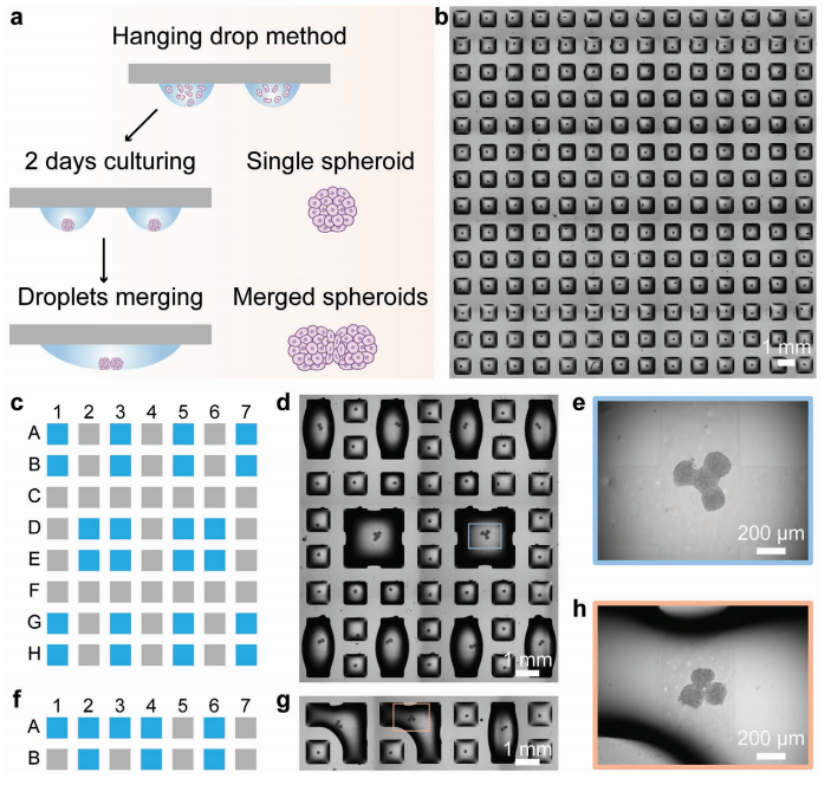
图1. 可编程液滴融合技术(proMAD)。a)利用亲水点被超疏水边界分隔的小型化液滴微阵列(DMA)形成细胞球体阵列的示意图。为了实现球体的可编程装配,通过增加相邻液滴的体积来合并液滴。b)单个HepG2球体14 × 14阵列显微镜图像。c, f)多液滴在DMA上可控合并的打印方案。使用以下印刷体积:900 nL /液滴,用于融合2或4个液滴;850 nL /液滴,将3个液滴融合在一起((c)和(f)中的蓝色点)。d, g)含有2、3和4个合并液滴的DMA显微镜图像。e, h)融合24 h后融合水滴中的4个球体和3个球体图像
1. 球体特征分析:使用ImageJ软件测量和计算球体的圆形度、纵横比、圆度和密实度等参数[5];
2. 融合过程分析:通过ImageJ软件的角度工具测量两个融合球体在不同融合时间点的夹角,用长度测量工具测量两个球体之间融合区域(“颈部”)的长度[5];
3. 细胞活力检测:融合后24 h收集双球体,用 PBS 洗涤后,用 Calcein- AM和碘化丙啶染色15 min(37°C),然后通过荧光显微镜成像检测细胞活力[5];
4. 免疫染色:收集并洗涤球体后,用 4% 多聚甲醛固定,0.5% Triton X - 100 透化,1% BSA 封闭,依次加入抗 - E - 钙粘蛋白抗体和 Alexa Fluor 488 标记的二抗,最后用 DAPI 染色,通过共聚焦显微镜成像[5];
5. 扫描电镜固定:融合后 24 h 收集球体,用 PBS 洗涤,依次用 2.5% 戊二醛溶液固定、不同浓度乙醇和水脱水、50% 和 100% HMDS 处理,然后转移到干净玻片上干燥,通过扫描电子显微镜成像[5]。
6. 球体间Wnt信号传导:产生HEK 293T Wnt-3a,以及稳定的HEK 293T TOP-GFP报告细胞系按照规划程序进行播种[6]。将产生Wnt的细胞打印在第1、3、5、7、9、11、13列,报告细胞打印在第2、4、6、8、10、12、14列。挂滴法培养2天后,每个点加入900 nL DMEM合并相邻点。为了研究Wnt信号传导,每24小时用荧光显微镜和共聚焦显微镜检查合并的球体。
1、对于融合的球体,定义了 “结合数” 来描述单个球体在融合过程中与相邻球体的作用。在这个过程中,I-DOT对前期球体的精准分配影响了后续融合的球体的各种数据。通过模拟算法和实验结果统计量化了合并球体 “结构异构体” 的组合概率。例如,3个球体融合时有两种可能的结构,线性组合A1A2A1和三角形结构A2A2A2,实验概率分别为61% 和39%,模拟算法得到的概率分别为85%和15%(图2a);
除了融合可变数量的同源球体外,还可以融合由不同细胞类型形成的球体(图2b)。在多细胞生物中,组织由多种具有空间组织的细胞类型组成,这些细胞类型协同工作以执行特定功能。图2b的结果显示,即使在融合过程24小时后,异质球体中两种细胞类型之间仍有明确的边界。因此,proMAD方法可以通过合并特定数量的相邻液滴来实现单个球体的可控组装,从而产生多球体聚集体,以及阵列格式的异质球体架构。
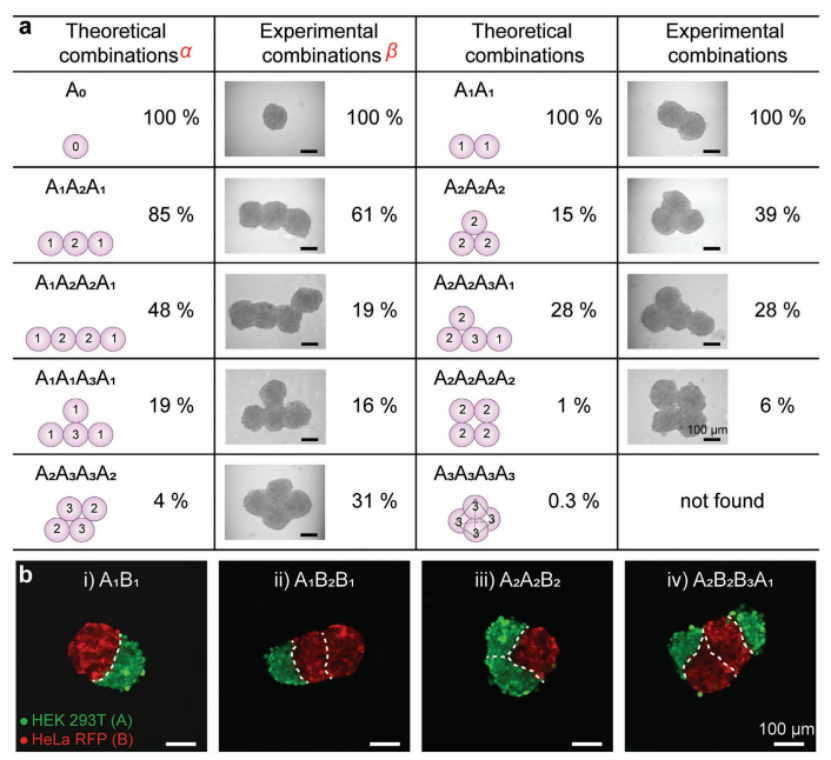
图2. 由proMAD方法形成的多球体结构的例子。a)不同HepG2多球体的示意图、概率和相应的例子。“结合数”对应于配合物的各个球体之间的结合位点的数量,显示在方案上的每个球体中。α,在理论模型中,将细胞球体视为球体,并通过模拟算法给出组合概率。β,组合概率由实验统计得出(3个球体,n = 33, 4个球体,n = 32)。b)两种不同细胞系(表达RFP和HEK 293T的HeLa细胞用绿色荧光5-氯甲基荧光素双乙酸酯染色)合并后24小时构建的异球结构的荧光显微镜图像:A1B1,A1B2B1,A2A2B2和A2B2B3A1。
2、为了观察球体融合的过程,我们在液滴融合后的5天内,在规定的时间点用显微镜监测了两个球体融合在一个液滴中的情况(图3a)。两个HepG2球体首先融合成花生状结构,水滴融合后24-96 h形成固体融合椭圆形结构(图3a)。在观察两个球体融合过程中,测量了融合点的切线角度和融合区域的长度。融合2 h后,两个原始球体的形状仍可清晰辨认,夹角在57° ± 16°到154° ± 11°之间,融合区域长度约为40.7 ± 6.8 µm;48 h后夹角大于,96 h完全融合时夹角接近,融合区域长度逐渐增加到138 ± 10µm(图3c),共培养48 h后,合并后96 h,两个球体之间的夹角在110°以上,逐渐接近180°,完全融合(图3a,b)。我们称之为“颈部”的合并区域的长度(图3a,橙色虚线)。
此外,作者还使用细胞-细胞相互作用模型来模拟两个球体的融合过程(图3d)[7]。热图(图3e)显示了融合过程中单个细胞的相对位移,表明球体中的细胞已经重新分布,形成新的椭圆形结构。在我们的实验和模拟中,融合后96 h双球体的总长度分别减少了约18%。
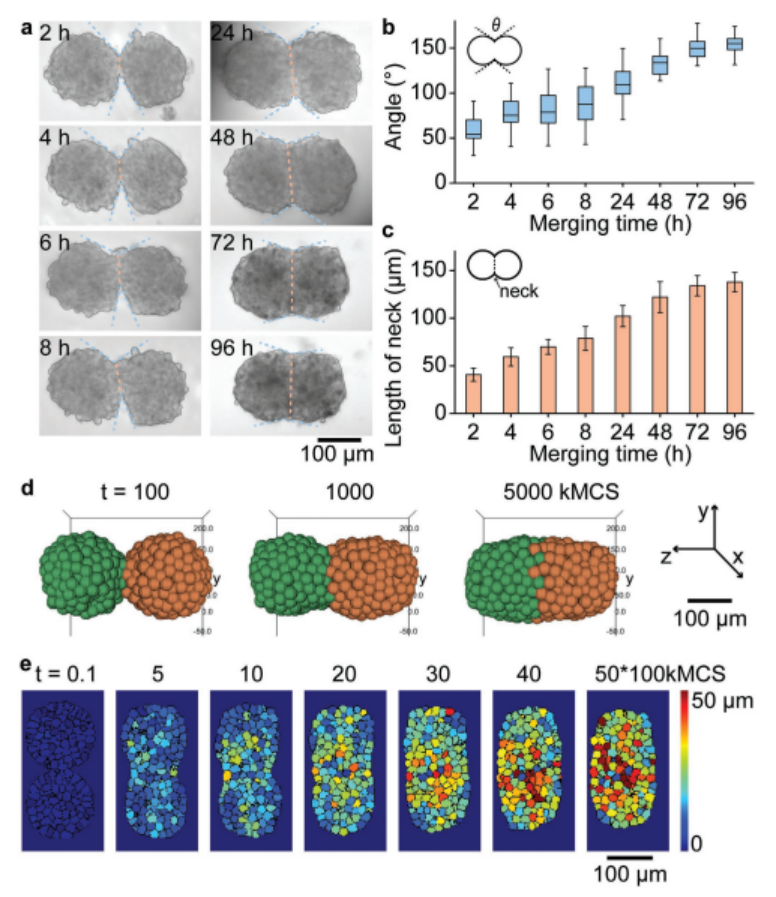
图3. 两胞球体融合过程的实验与模拟研究。a)两个HepG2球体初次接触96小时后融合的亮场显微镜图像。b)合并球体间夹角随时间变化的曲线图。(a)中的蓝色虚线表示角度。c)显示合并球体随时间变化的颈部长度(橙色虚线显示在(a)中)的图。(b)和(c)中的误差条表示10个不同合并球体的SD。d)两个球体(球体直径150µm, 400个细胞)融合过程的三维模拟。e)热图显示融合过程中单个细胞的时空运动。彩色地图表示每个细胞相对于其初始位置的位移(t = 0)。kMCS是时间尺度,代表kilo Monte Carlo sweep。
3、为观察两球体合并区域细胞状况,进行了E-cadherin免疫染色和扫描电镜成像(图4a)。E-cadherin免疫染色用于可视化细胞粘附和连接[8],结果显示在融合的双球体内均匀分布,表明球体是生物融合而非物理吸附。扫描电镜成像证实了 “花生” 形融合复合体的形成及单个球体界面细胞间的紧密相互作用(图4b)。这些结果表明,用特定方法获得的多球体三维结构,在球体之间具有紧密的细胞间接触,利于研究细胞间相互作用及相关生物过程。

图4. 两胞球体融合过程的实验与模拟研究。多球体结构中的细胞-细胞相互作用。a)融合24 h后两个HepG2球体的荧光显微镜图像。e -钙粘蛋白染色(绿色荧光)显示细胞间连接。DAPI(蓝色荧光)显示细胞核。合并:DAPI覆盖层,E-Cadherin染色。b)融合后球体(HepG2,融合后24 h)的SEM图像。左图是在2000倍放大率下拍摄的。右上方图像(蓝框)在7000倍放大下显示单个球体内细胞-细胞接触。右下角(橙色框)在7000倍放大下显示了双球体的接触“颈部”区域。
4、使用proMAD方法将产生Wnt的球体(HEK 293T, Wnt-3a)与报告球体(HEK 293T, TOP-GFP)融合(图5a)。融合后24和48小时,通过测量TOP-GFP报告球体的荧光强度来监测Wnt信号活性(图5b)。报告球体共培养24 h后平均荧光强度为45.3 ± 10.0,共培养48 h后平均荧光强度为58.3 ± 5.7(图5b)。共培养48小时后,整个报告球体显示出均匀的GFP荧光,表明在球体的所有细胞中Wnt信号被强烈激活(图5c-i,ii)。对照实验仅显示较弱的GFP强度(图5c-i,ii) [9,10],融合后24和48 h的平均荧光强度分别为3.0 ± 1.1和2.9 ± 0.9。在Wnt-3a存在下,TOP-GFP Wnt报告球体的荧光强度比对照球体增加了约15倍(24 h)和大约20倍(48 h)。
接下来,研究测试了Wnt信号在三重球体复合体中的传播,将两个报告球体与一个产生Wnt-3a的球体合并,然后选择两种不同的融合球体架构进行分析。在第一种结构中,两个报告球体呈线性结构排列,产生Wnt的球体位于一端(图5d)。与产生Wnt直接相邻的报告球体被强烈激活,而位于远端的报告球体被不那么强烈激活(图5d,f)。在第二种组合中,两个报告球体位于生产者球体的两侧,显示出两个报告球状体在同等程度上的强烈激活(图5e,g)
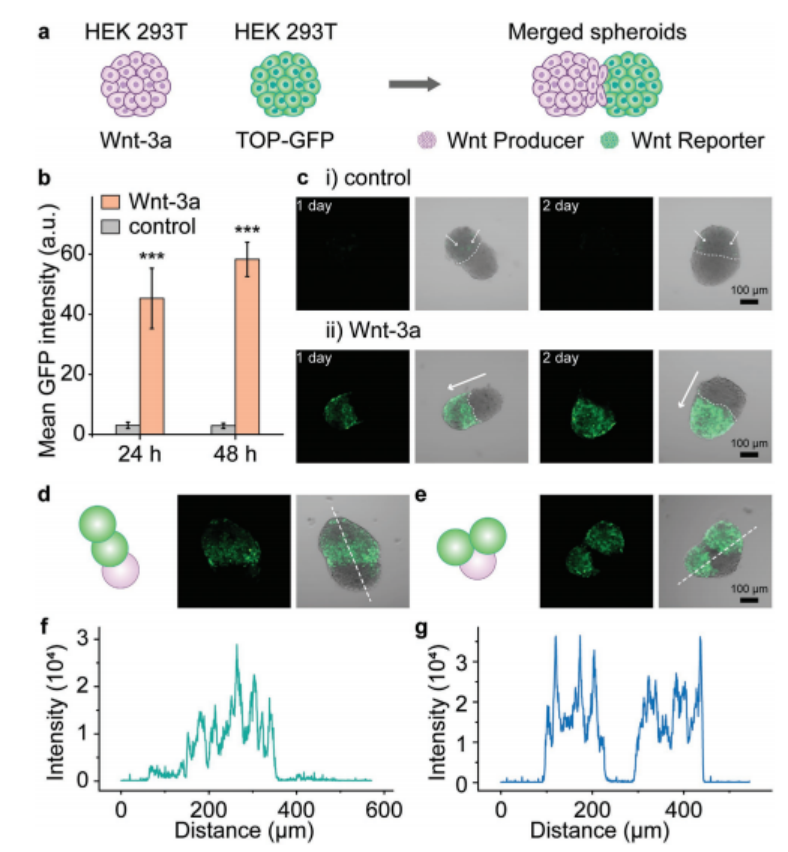
图5. 合并球体之间的Wnt信号。a)由产生球体(HEK 293T、Wnt-3a)和报告球体(HEK 293T、TOP-GFP)组成的多球体复合物构成的Wnt信号传播系统示意图。b)报告球体激活图。通过共聚焦图像中GFP荧光的强度来估计激活。(GFP强度由至少10个合并的球体计算。***, P < 0.001,单因素方差分析)c)共聚焦图像显示GFP荧光估计报告球体中Wnt/β-catenin信号的激活。i) HEK 293T球体(对照)和报告球体的合并球体。白色箭头指向微弱的GFP荧光。ii)一个Wnt产生球(HEK 293T)和一个Wnt报告球(HEK 293T, TOP-GFP)的合并球。白色箭头指向Wnt传播方向。d,e)所研究的两个三重球体的示意图和荧光显微镜图像,其中一个Wnt产生球体(HEK 293T)和两个Wnt报告球体(HEK 293T, TOP-GFP)。f)如图(d)所示三重球体白色虚线上GFP荧光强度分布图。g)如图(e)所示三重球体白色虚线上GFP荧光强度分布图。
本文通过proMAD方法成功实现了多个同型或异型球体的融合,形成了双球体、多球体和异质球体等各种多球体结构,并确定了不同结构的组合概率。与现有的3D细胞培养方法相比,proMAD方法具有诸多独特优势。如高通量、小型化、已操作及快速形成复杂多极结构等。I-DOT在前期球体的制备过程中保证了每个液滴的准确性,间接影响了最终多球体结构的形成。同时,该方法也为研究3D细胞培养模型中的细胞信号传导提供了强大工具,I-DOT在多个关键步骤中发挥重要作用的同时,更有助于在更接近生理条件下研究各种生物学过程,为组织工程和再生医学的发展提供新的思路和方法。
同腾睿杰(上海)生物技有限公司作为CYTENA I.DOT中国总代理商,为您提供优质的售前售后服务。
联系电话:021-50826962
联系邮箱:sales@ttbiotech.com


